
{kind=link}
Protenix-v1 — Key Takeaways
- Protenix-v1 replicates AlphaFold3 performance for diverse biomolecular structures.
- Released under Apache 2.0, it includes complete code and parameters.
- The PXMeter toolkit enables benchmarking over 6,000 complexes for transparency.
- Outperforms AlphaFold3 while maintaining identical training data conditions.
- Enhanced accuracy for hard targets is achieved by sampling more candidates.
What We Know So Far
ByteDance’s New Model
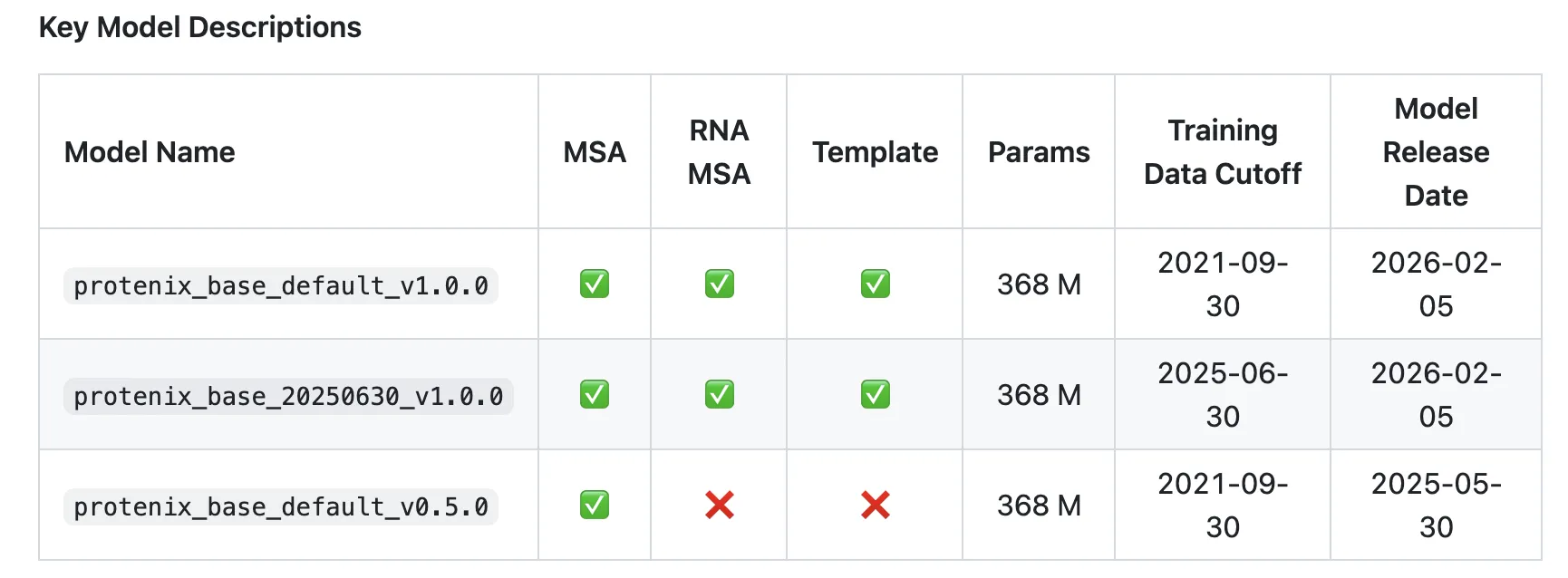
ByteDance has launched Protenix-v1, a sophisticated open-source model for biomolecular structure prediction. This initiative aims to mirror the groundbreaking performance of AlphaFold3, particularly across various biomolecular structures such as proteins, DNA, RNA, and ligands. By focusing on accessibility and transparency, ByteDance encourages wider utilization in the scientific community.
Related image — Source: marktechpost.com — Original
This model embodies significant innovation and represents an advancement in computational biology, specifically designed to provide cutting-edge tools for researchers and practitioners in the field. The model’s underlying technology leverages years of development and research in machine learning and biomolecular modeling.
Released under the Apache 2.0 license, Protenix-v1 includes a full codebase along with model parameters, allowing researchers and developers to utilize it for both academic and commercial purposes. This open-source approach not only democratizes access to high-quality modeling tools but also fosters collaboration and the sharing of ideas among scientists.
Key Details and Context
To better understand Protenix-v1, it’s crucial to consider its context within the field of biomolecular modeling. The continued advancements in machine learning have propelled a renaissance in protein structure prediction, leading to more accurate and efficient models. Protenix-v1 stands at the forefront of this trend.

Related image — Source: marktechpost.com — Original
More Details from the Release
PXMeter provides a clear and documented inference-time scaling behavior that differs from a single fixed operating point during predictions. This feature is particularly beneficial for researchers needing reliable performance metrics when evaluating various protein folding scenarios.
The research indicates that increasing the number of sampled candidates for challenging targets such as antigen-antibody complexes significantly improves accuracy. This insight is pivotal, as it allows researchers to strategically increase sampling in experiments involving these complex interactions.
Protenix-v1 is described as outperforming AlphaFold3 across diverse benchmark sets while adhering to the same training data cutoff and model scale. Such achievements mark an essential milestone in the ongoing race for better predictive accuracy in the field.
Additionally, Protenix-v1 re-implements the AF3-style diffusion architecture for all-atom complexes using a trainable PyTorch codebase. This choice not only enhances user experience but also aligns with the widely adopted ecosystem of machine learning, contributing to community engagement.
Importantly, Protenix-v1 includes an evaluation toolkit named PXMeter v1.0.0 which allows transparent benchmarking across more than 6,000 complexes. This extensive benchmarking capability underscores the model’s reliability and effectiveness in various applications.
As a result, Protenix-v1 aims to achieve AlphaFold3-level performance across various biomolecular structures including proteins, DNA, RNA, and ligands. ByteDance has introduced this model as a comprehensive reproduction of AlphaFold3 for biomolecular structure prediction.
Evaluation with PXMeter
One of the standout features of Protenix-v1 is PXMeter v1.0.0, an evaluation toolkit that enables transparent benchmarking across a library of over 6,000 biomolecular complexes. This tool is designed for clear evaluations, allowing users to assess model performance conveniently.
Notably, Protenix-v1 re-implements the AF3-style diffusion architecture for all-atom complexes using a trainable PyTorch codebase, which enhances accessibility for researchers familiar with this environment. This approach helps bridge the gap between complex scientific research and practical implementation, making it easier for users to contribute to the model’s development.
What Happens Next
Benchmarking Against AlphaFold3
Research indicates that Protenix-v1 consistently outperforms AlphaFold3 across various benchmark sets, while adhering to the same training data cutoff and model scale. This positions Protenix-v1 as a robust alternative, inspiring further advances in biomolecular modeling.

Related image — Source: marktechpost.com — Original
Additionally, the researchers found that increasing the number of sampled candidates for challenging targets, like antigen-antibody complexes, can significantly improve accuracy, a critical factor in real-world applications. Such fine-tuning is essential for practical scenarios where prediction accuracy can directly impact experimental outcomes.
Why This Matters
Impact on Biomolecular Research
The introduction of Protenix-v1 represents a notable advancement in the field of biomolecular structure prediction. Its open-source nature democratizes access to cutting-edge technology, fostering innovation and collaboration among researchers globally. A diverse array of scientific professionals can now explore and adapt the technology to their specific needs.
As a tool that not only equals but often exceeds AlphaFold3’s capabilities, Protenix-v1 could lead to breakthroughs in drug discovery and therapeutic development by providing accurate predictions of complex biological structures. This capability is crucial in advancing the understanding and treatment of various diseases, enhancing the potential for breakthroughs in clinical trials and related fields.
FAQ
Common Questions About Protenix-v1
Protenix-v1’s release has generated many inquiries from the research community. Here are some key questions that have arisen:
Protenix-v1 is free to use, and its versatile capabilities make it suitable for modeling various biomolecules, including proteins, DNA, RNA, and ligand structures. Its user-friendly nature invites not only seasoned researchers but also newcomers in the field to leverage its functionalities.